Introduzione all’analisi dei dati RNA-Seq
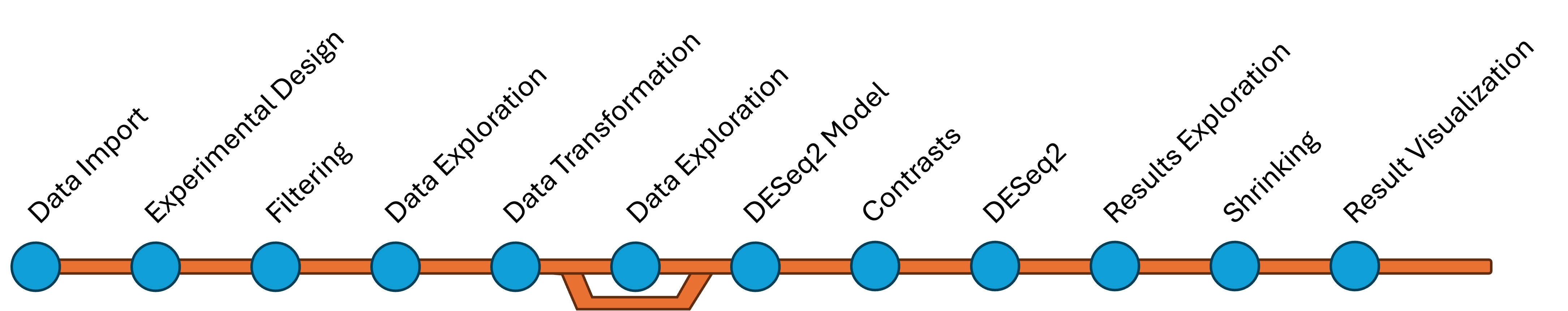
Benvenuti nel complesso mondo dell’analisi dei dati di sequenziamento RNA! Come illustrato nella Figure 1, il processo di analisi si articola in diverse fasi cruciali e interconnesse. Ogni step si fonda sui risultati del precedente, permettendoci di estrarre informazioni biologiche significative dai dati grezzi generati dagli esperimenti di RNA-seq.
La Figure 1 rappresenta una roadmap per la nostra analisi. Inizieremo con l’importazione e la pre-elaborazione dei dati di conteggio, provenienti da strumenti di quantificazione (HTSeq)o quasi-mapping (Salmon). Successivamente, utilizzeremo DESeq2 per modellare i dati di conteggio, tenendo conto del disegno sperimentale e delle caratteristiche intrinseche dei dati RNA-seq, come la variabilità inter-replicato e la sovra-dispersione. L’obiettivo finale è identificare i geni differenzialmente espressi tra le diverse condizioni sperimentali, interpretando i risultati in un contesto biologico.”
Introduzione dettagliata di ogni fase:
Importazione dei Dati: “Il punto di partenza è l’importazione dei dati grezzi di conteggio, tipicamente organizzati in una matrice geni x campioni. Oltre ai conteggi genici, potremmo dover gestire dati di abbondanza a livello di trascritto, sfruttando pacchetti come
tximportotximetaper l’integrazione con DESeq2. È fondamentale assicurare la corretta formattazione dei dati e la compatibilità con il pacchetto DESeq2.”Disegno Sperimentale: “La definizione del disegno sperimentale è un prerequisito fondamentale per un’analisi accurata. Dovremo costruire una tabella di metadati che annoti in modo preciso ogni campione, specificando i fattori sperimentali (es. condizioni di trattamento, tessuti, tempi) e i loro livelli. Queste informazioni saranno integrate nel modello statistico per l’analisi dell’espressione differenziale.”
Filtraggio: “Il filtraggio dei geni con basso conteggio è un passaggio importante per migliorare l’affidabilità dell’analisi. Geni con un numero insufficiente di reads mappate possono introdurre rumore e distorcere i risultati. Possiamo applicare criteri di filtraggio basati su soglie di conteggio o sull’informazione di annotazione genica.”
Esplorazione dei Dati (Conteggi Grezzi): “Prima di applicare qualsiasi trasformazione, è essenziale analizzare i dati di conteggio grezzi. Attraverso la visualizzazione delle distribuzioni, l’analisi delle relazioni tra campioni (es. PCA) e l’identificazione di potenziali outlier, possiamo valutare la qualità dei dati e individuare eventuali problematiche che richiedono attenzione.”
-
Trasformazione dei Dati: “Per stabilizzare la varianza e rendere i dati più adatti alle assunzioni del modello statistico, possiamo applicare trasformazioni come la Variance Stabilizing Transformation (VST) o la Regularized Log Transformation (rlog). La scelta della trasformazione dipenderà dalle caratteristiche dei dati e dagli obiettivi dell’analisi.”
-
VST vs. rlog
Sia la VST (Variance Stabilizing Transformation) che la rlog (Regularized Log Transformation) mirano a trasformare i dati di conteggio per stabilizzare la varianza tra i geni con diversa espressione. Questo è importante perché il modello statistico di DESeq2 assume che la varianza sia approssimativamente costante per tutti i geni.
Quando scegliere VST?
- Analisi di Espressione Differenziale: La VST è generalmente preferita per l’analisi dell’espressione differenziale, poiché è stata specificamente progettata per questo scopo.
- Visualizzazione di Heatmap e PCA: La VST è adatta per visualizzare i dati in heatmap e PCA, in quanto preserva bene le distanze tra i campioni.
- Dataset di Grandi Dimensioni: La VST è computazionalmente più efficiente della rlog, quindi è preferibile per dataset con molti geni o campioni.
Quando scegliere rlog?
- Esplorazione dei Dati: La rlog può essere utile per esplorare i dati in modo più intuitivo, poiché produce valori che sono più simili ai log2 fold change.
- Clustering: La rlog può essere preferita per il clustering dei geni, poiché tende a separare meglio i cluster.
- Dataset di Piccole Dimensioni: La rlog può essere più accurata della VST per dataset con pochi geni o campioni.
-
Esplorazione dei Dati (Dati Trasformati): “Dopo la trasformazione, è opportuno riesaminare i dati per valutare l’effetto della trasformazione e confrontarli con i conteggi grezzi. Questo ci permette di verificare l’efficacia della trasformazione e di identificare eventuali artefatti.”
Modello DESeq2: “Il cuore dell’analisi risiede nel modello statistico implementato in DESeq2. Questo modello si basa sulla distribuzione binomiale negativa, che è particolarmente adatta per modellare i dati di conteggio RNA-seq, caratterizzati da sovra-dispersione. DESeq2 stima i fattori di normalizzazione per le dimensioni della libreria e la dispersione genica, parametri cruciali per l’identificazione dei geni differenzialmente espressi.”
Contrasti: “Per effettuare confronti specifici tra le condizioni sperimentali, dobbiamo definire i contrasti di interesse. I contrasti specificano quali gruppi confrontare e la direzione dell’effetto atteso. DESeq2 consente di definire contrasti complessi, includendo interazioni tra fattori e analisi di serie temporali.”
DESeq2: “In questa fase, applichiamo la funzione
DESeq()per eseguire l’analisi dell’espressione differenziale. DESeq2 utilizza il modello statistico e i contrasti definiti per calcolare i log2 fold change, i valori p e i valori p aggiustati per ogni gene.”Esplorazione dei Risultati: “L’analisi dei risultati consiste nell’esaminare le tabelle generate da DESeq2, individuando i geni differenzialmente espressi in base a criteri di significatività statistica (valori p aggiustati) e di rilevanza biologica (log2 fold change). È importante considerare sia la significatività che la dimensione dell’effetto.”
Shrinking: “Per ottenere stime più accurate e interpretabili dei log2 fold change, possiamo applicare metodi di shrinkage. Lo shrinkage riduce l’effetto dei geni con bassa espressione o alta varianza, migliorando l’affidabilità delle stime.”
Visualizzazione dei Risultati: “La visualizzazione dei risultati è fondamentale per la comunicazione e l’interpretazione dei dati. Utilizzeremo grafici come MA plot, volcano plot e heatmap per rappresentare i risultati dell’analisi in modo efficace, evidenziando i geni differenzialmente espressi e i pattern di espressione.”