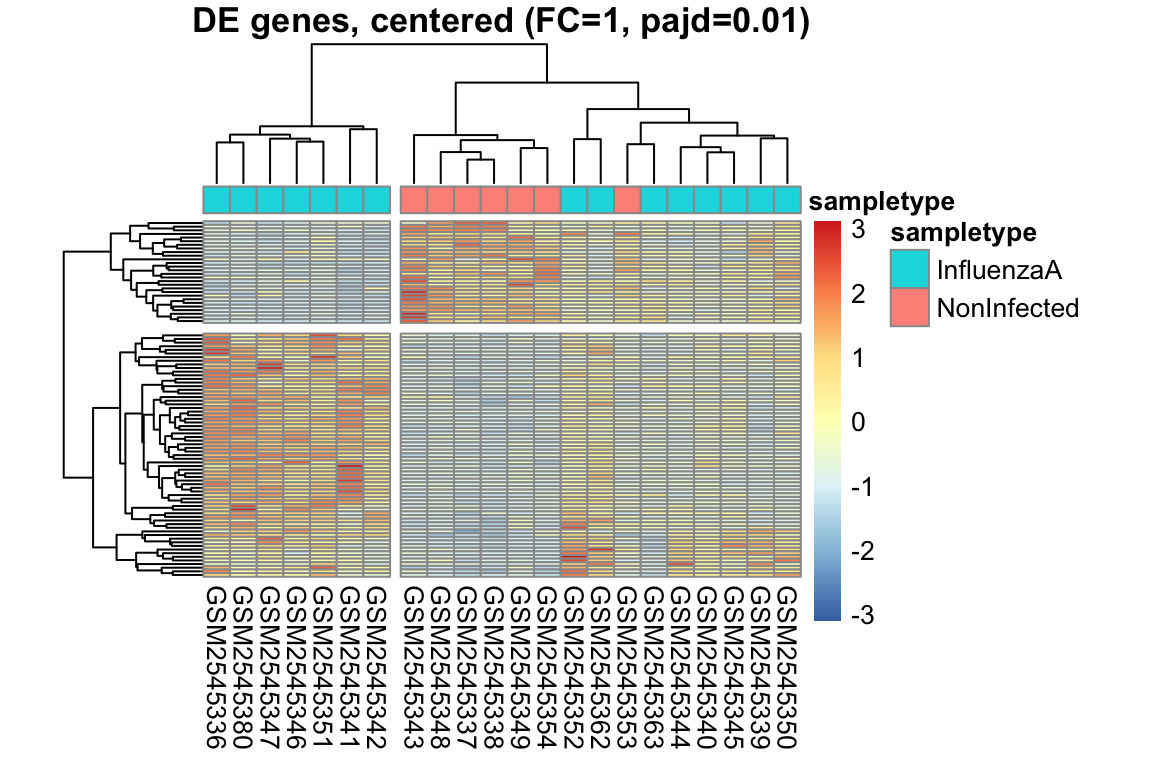
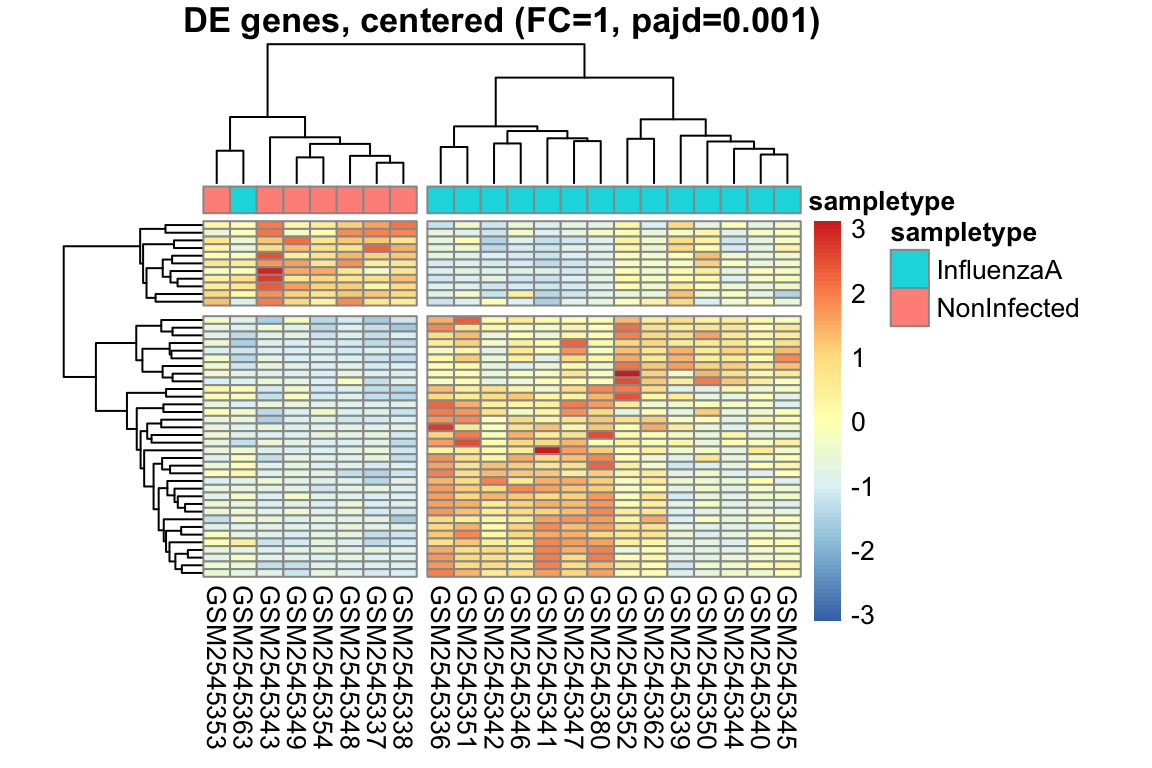
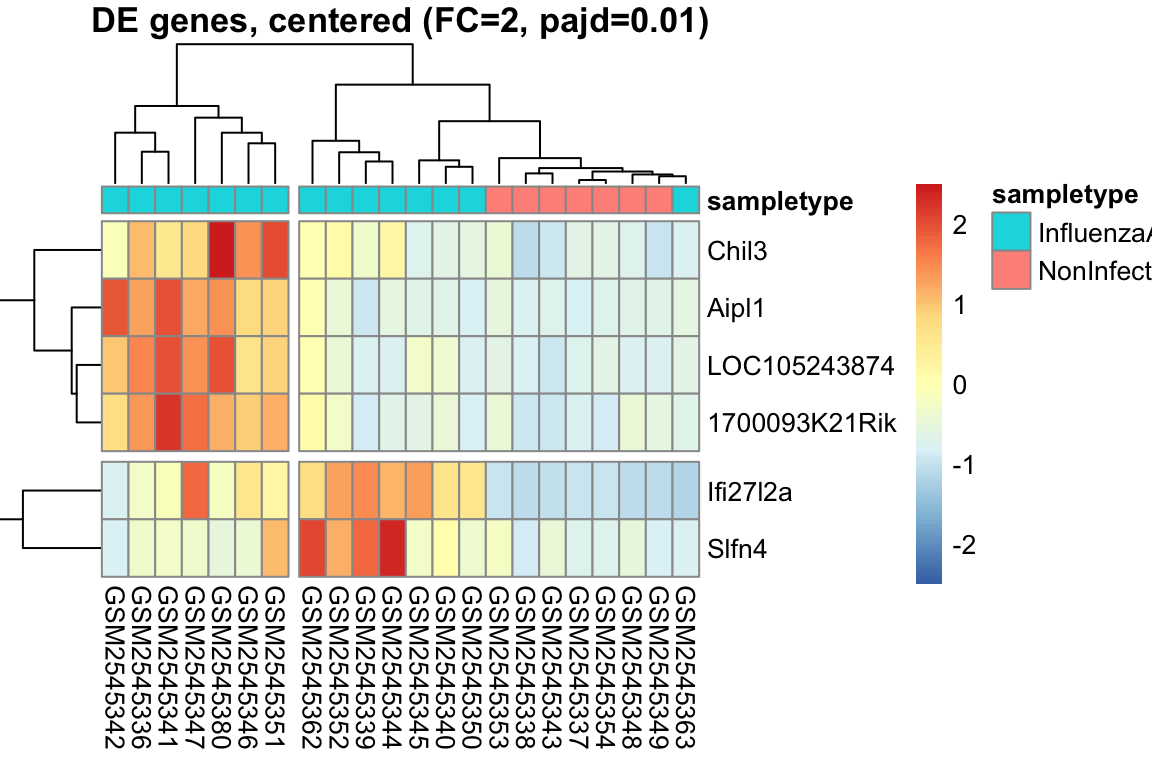
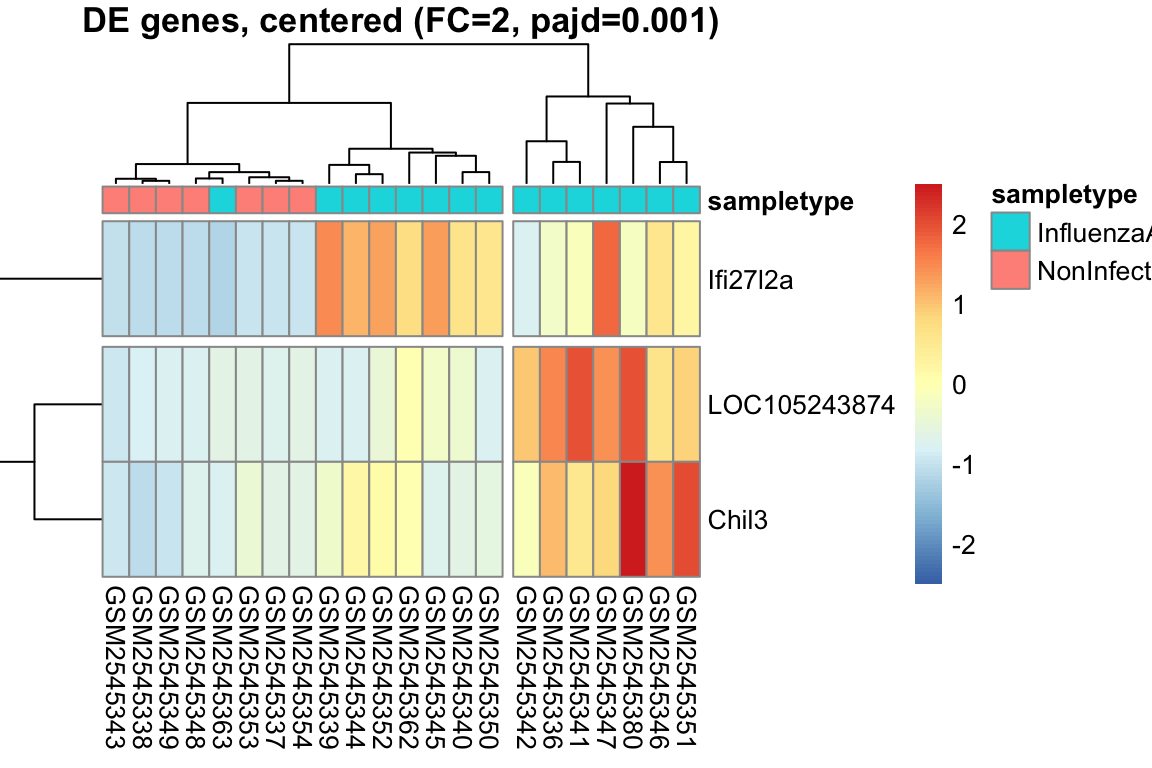
L’heatmap è una rappresentazione grafica a matrice che utilizza colori per visualizzare i valori di un set di dati. Nell’analisi dell’espressione genica, l’heatmap è uno strumento potente per visualizzare i pattern di espressione di più geni in diversi campioni simultaneamente.
Come funziona:
Organizzazione dei dati: I dati di espressione genica sono organizzati in una matrice, dove le righe rappresentano i geni e le colonne rappresentano i campioni.
Mappa di colori: I valori di espressione sono mappati a una scala di colori graduata. In genere, colori freddi (ad esempio, il blu) rappresentano bassi livelli di espressione, mentre colori caldi (ad esempio, il rosso) rappresentano alti livelli di espressione.
Visualizzazione: La matrice viene visualizzata come una griglia di celle colorate, dove ogni cella rappresenta il livello di espressione di un gene in un campione specifico.
Clustering gerarchico:
Spesso, gli heatmap vengono combinati con il clustering gerarchico, un metodo statistico che raggruppa geni e/o campioni con pattern di espressione simili.
Clustering dei geni: Geni con profili di espressione simili sono raggruppati insieme, visualizzati come righe adiacenti nell’heatmap.
Clustering dei campioni: Campioni con profili di espressione simili sono raggruppati insieme, visualizzati come colonne adiacenti nell’heatmap.
Vantaggi:
Visione d’insieme: Gli heatmap forniscono una panoramica completa dei pattern di espressione di un gran numero di geni in diversi campioni.
Identificazione di pattern: Permettono di identificare facilmente:
Geni co-espressi, ovvero geni con livelli di espressione simili in diversi campioni.
Gruppi di campioni con profili di espressione simili.
Outlier, ovvero geni o campioni con pattern di espressione insoliti.
Esplorazione di dataset complessi: Sono uno strumento efficace per esplorare set di dati di grandi dimensioni e complessi.
Intensità del colore: Indica il livello di espressione del gene nel campione.
Dendrogramma: Mostra le relazioni gerarchiche tra geni (righe) e campioni (colonne).
Pattern: Cerca blocchi di colore, che indicano gruppi di geni co-espressi o gruppi di campioni con profili simili.
Source Code
---params: mycondition: infection mynum: InfluenzaA mydenom: NonInfected mypval: 0.01 myfc: 0.8 mypadj: fdr---```{r}#| echo: false#| message: false#| warning: falsesource("_common.R")library(tidyverse)library(DESeq2) # BioClibrary(RColorBrewer)library(pheatmap)library(ggrepel)library(cowplot)library(DT)library(scales)library(vsn) # BioClibrary(apeglm) # BioClibrary(rmarkdown)library(gt)dds <-readRDS("data/DE.rds")res <-readRDS("data/res.rds")```# Heatmap plot {#sec-vis-heatmap}L'heatmap è una rappresentazione grafica a matrice che utilizza colori per visualizzare i valori di un set di dati. Nell'analisi dell'espressione genica, l'heatmap è uno strumento potente per visualizzare i pattern di espressione di più geni in diversi campioni simultaneamente.::: {.content-hidden when-meta="features.advanced_analysis"}**Come funziona:**- **Organizzazione dei dati:** I dati di espressione genica sono organizzati in una matrice, dove le righe rappresentano i geni e le colonne rappresentano i campioni.- **Mappa di colori:** I valori di espressione sono mappati a una scala di colori graduata. In genere, colori freddi (ad esempio, il blu) rappresentano bassi livelli di espressione, mentre colori caldi (ad esempio, il rosso) rappresentano alti livelli di espressione.- **Visualizzazione:** La matrice viene visualizzata come una griglia di celle colorate, dove ogni cella rappresenta il livello di espressione di un gene in un campione specifico.**Clustering gerarchico:**Spesso, gli heatmap vengono combinati con il clustering gerarchico, un metodo statistico che raggruppa geni e/o campioni con pattern di espressione simili.- **Clustering dei geni:** Geni con profili di espressione simili sono raggruppati insieme, visualizzati come righe adiacenti nell'heatmap.- **Clustering dei campioni:** Campioni con profili di espressione simili sono raggruppati insieme, visualizzati come colonne adiacenti nell'heatmap.**Vantaggi:**- **Visione d'insieme:** Gli heatmap forniscono una panoramica completa dei pattern di espressione di un gran numero di geni in diversi campioni.- **Identificazione di pattern:** Permettono di identificare facilmente: - Geni co-espressi, ovvero geni con livelli di espressione simili in diversi campioni. - Gruppi di campioni con profili di espressione simili. - Outlier, ovvero geni o campioni con pattern di espressione insoliti.- **Esplorazione di dataset complessi:** Sono uno strumento efficace per esplorare set di dati di grandi dimensioni e complessi.:::## FC=1 & padj=0.01```{r}#| echo: true#| fig-width: 6#| fig-height: 16#| fig-align: centerhm <-function(res, pd =0.001, lFC =1, gene =FALSE) { resSubset <-subset(res, padj < pd &abs(log2FoldChange) > lFC) resSubset <- resSubset[order(resSubset$log2FoldChange, decreasing =TRUE), ] genesSelected <-rownames(resSubset) heatData <-assay(dds)[genesSelected, ] my_sample_col <-as.data.frame(cbind(rownames(colData(dds)), colData(dds)["infection"]))colnames(my_sample_col) <-c("name", "sampletype")rownames(my_sample_col) <- my_sample_col$name my_sample_col$name <-NULLpheatmap(heatData, annotation_col = my_sample_col, cellwidth =10, scale ="row", cluster_rows =TRUE, cluster_cols =TRUE, show_rownames = gene, main =paste0("DE genes, centered (FC=", lFC, ", pajd=", pd, ")"), cutree_rows =2, cutree_cols =2)}hm(res, 0.01, 1)```## FC=1 & padj=0.001```{r}#| echo: truehm(res, 0.001, 1)```## FC=2 & padj=0.01 & gene names```{r}#| echo: truehm(res, 0.01, 2, TRUE)```## FC=2 & padj=0.001 & gene names```{r}#| echo: truehm(res, 0.001, 2, TRUE)```**Interpretazione:**- **Intensità del colore:** Indica il livello di espressione del gene nel campione.- **Dendrogramma:** Mostra le relazioni gerarchiche tra geni (righe) e campioni (colonne).- **Pattern:** Cerca blocchi di colore, che indicano gruppi di geni co-espressi o gruppi di campioni con profili simili.