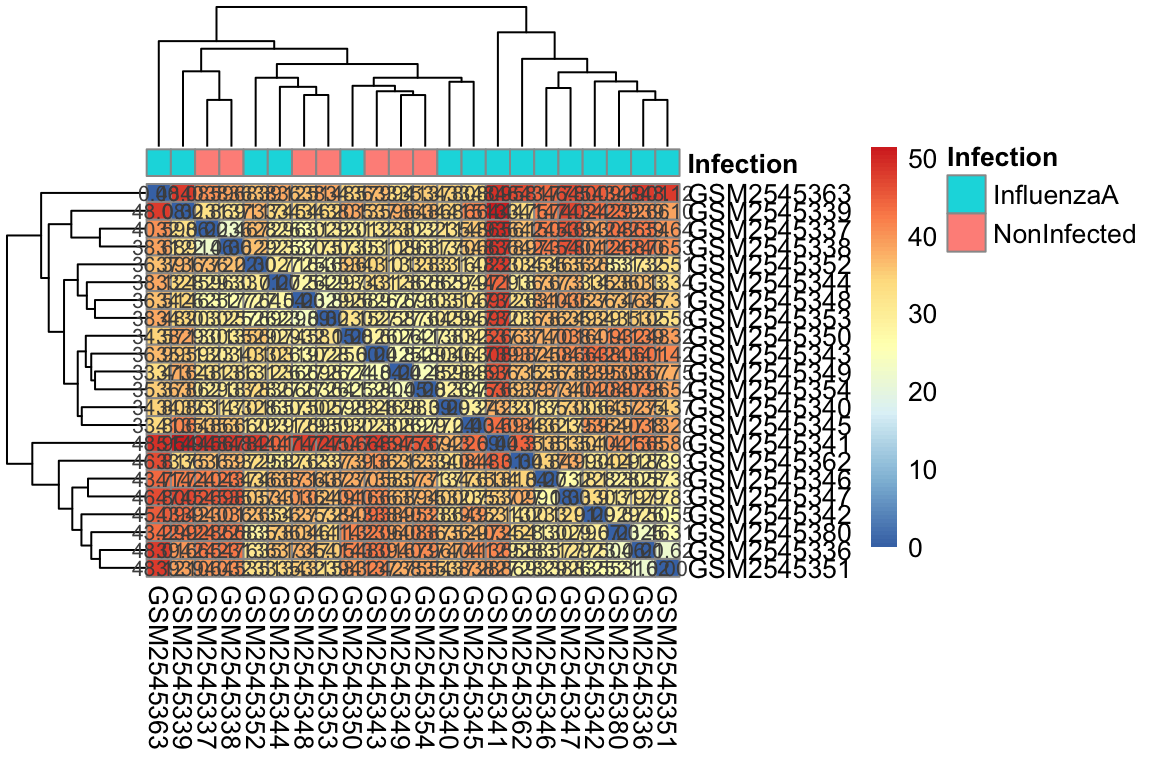
Un heatmap è uno strumento potente per visualizzare dati di espressione genica, in quanto consente di osservare pattern di espressione di più geni in diversi campioni simultaneamente.
Come funziona:
Matrice di dati: I dati di espressione genica sono organizzati in una matrice, dove le righe rappresentano i geni e le colonne rappresentano i campioni. Ogni cella della matrice contiene un valore che indica il livello di espressione di un particolare gene in un particolare campione.
Mappa di colori: I valori di espressione sono mappati a una scala di colori, in genere con colori freddi (ad esempio, blu) che rappresentano bassi livelli di espressione e colori caldi (ad esempio, rosso) che rappresentano alti livelli di espressione.
Visualizzazione: La matrice viene visualizzata come una griglia di celle colorate, dove ogni cella rappresenta un valore di espressione. I pattern di espressione possono essere facilmente identificati osservando le variazioni di colore nella griglia.
Clustering gerarchico:
Spesso, i heatmap vengono combinati con il clustering gerarchico per raggruppare geni e campioni con pattern di espressione simili.
Clustering dei campioni: I campioni con pattern di espressione simili vengono raggruppati insieme in colonne.
pcaData<-DESeq2::plotPCA(vst, intgroup =c("infection"), ntop =3000)#> using ntop=3000 top features by variancepcaData2<-pcaData$datapp<-ggplot(pcaData2, aes(x =PC1, y =PC2, text =name))+geom_point(aes(color =infection), size =5)+theme_minimal()plotly::ggplotly(pp)
6.3 Esplorazioni interattiva dei dati
Mostra il codice R
## Convert DESeqDataSet object to a SingleCellExperiment object, in order to## be able to store the PCA representation# library(SingleCellExperiment)# sce <- as(dds, "SingleCellExperiment")# ## Add PCA to the 'reducedDim' slot# stopifnot(rownames(pcaData) == colnames(sce))# reducedDim(sce, "PCA") <- as.matrix(pcaData[, c("PC1", "PC2")])# ## Add variance-stabilized data as a new assay# stopifnot(colnames(vst) == colnames(sce))# assay(sce, "vst") <- assay(vst)# app <- iSEE::iSEE(sce)# shiny::runApp(app)
Source Code
---params: mycondition: infection mynum: InfluenzaA mydenom: NonInfected mypval: 0.01 myfc: 0.8 mypadj: fdr---```{r}#| echo: false#| message: false#| warning: falsesource("_common.R")library(tidyverse)library(DESeq2) # BioClibrary(RColorBrewer)library(pheatmap)library(ggrepel)library(cowplot)library(DT)library(scales)library(vsn) # BioClibrary(apeglm) # BioClibrary(rmarkdown)library(gt)readcounts <-readRDS("data/readcounts.rds")coldata <-readRDS("data/coldata.rds")dds <-readRDS("data/dds_fitered.rds")vst <-readRDS("data/vst.rds")```# Esplorazione dei Dati {#sec-preproc-edatrans}## HeatmapUn heatmap è uno strumento potente per visualizzare dati di espressione genica, in quanto consente di osservare pattern di espressione di più geni in diversi campioni simultaneamente.::: {.content-hidden when-meta="features.advanced_analysis"}**Come funziona:**1. **Matrice di dati:** I dati di espressione genica sono organizzati in una matrice, dove le righe rappresentano i geni e le colonne rappresentano i campioni. Ogni cella della matrice contiene un valore che indica il livello di espressione di un particolare gene in un particolare campione.2. **Mappa di colori:** I valori di espressione sono mappati a una scala di colori, in genere con colori freddi (ad esempio, blu) che rappresentano bassi livelli di espressione e colori caldi (ad esempio, rosso) che rappresentano alti livelli di espressione.3. **Visualizzazione:** La matrice viene visualizzata come una griglia di celle colorate, dove ogni cella rappresenta un valore di espressione. I pattern di espressione possono essere facilmente identificati osservando le variazioni di colore nella griglia.**Clustering gerarchico:**Spesso, i heatmap vengono combinati con il clustering gerarchico per raggruppare geni e campioni con pattern di espressione simili.- **Clustering dei campioni:** I campioni con pattern di espressione simili vengono raggruppati insieme in colonne.:::```{r}#| out-width: 100%dst <-as.matrix(dist(t(assay(vst)))) |>data.frame()acol <-data.frame("Infection"= vst$infection)arow <-data.frame("Sex"= vst$infection)rownames(acol) <- vst$geo_accessionrownames(arow) <- vst$geo_accessionpheatmap(dst, annotation_col = acol, cluster_row =TRUE, display_numbers =TRUE)```## PCA```{r}#| out-width: 100%pcaData <- DESeq2::plotPCA(vst, intgroup =c("infection"), ntop =3000)pcaData2 <- pcaData$datapp <-ggplot(pcaData2, aes(x = PC1, y = PC2, text = name)) +geom_point(aes(color = infection), size =5) +theme_minimal() plotly::ggplotly(pp)```## Esplorazioni interattiva dei dati```{r}## Convert DESeqDataSet object to a SingleCellExperiment object, in order to## be able to store the PCA representation# library(SingleCellExperiment)# sce <- as(dds, "SingleCellExperiment")# ## Add PCA to the 'reducedDim' slot# stopifnot(rownames(pcaData) == colnames(sce))# reducedDim(sce, "PCA") <- as.matrix(pcaData[, c("PC1", "PC2")])# ## Add variance-stabilized data as a new assay# stopifnot(colnames(vst) == colnames(sce))# assay(sce, "vst") <- assay(vst)# app <- iSEE::iSEE(sce)# shiny::runApp(app)```