Mostra il codice R
# Crea un MA-plot
plotMA(res)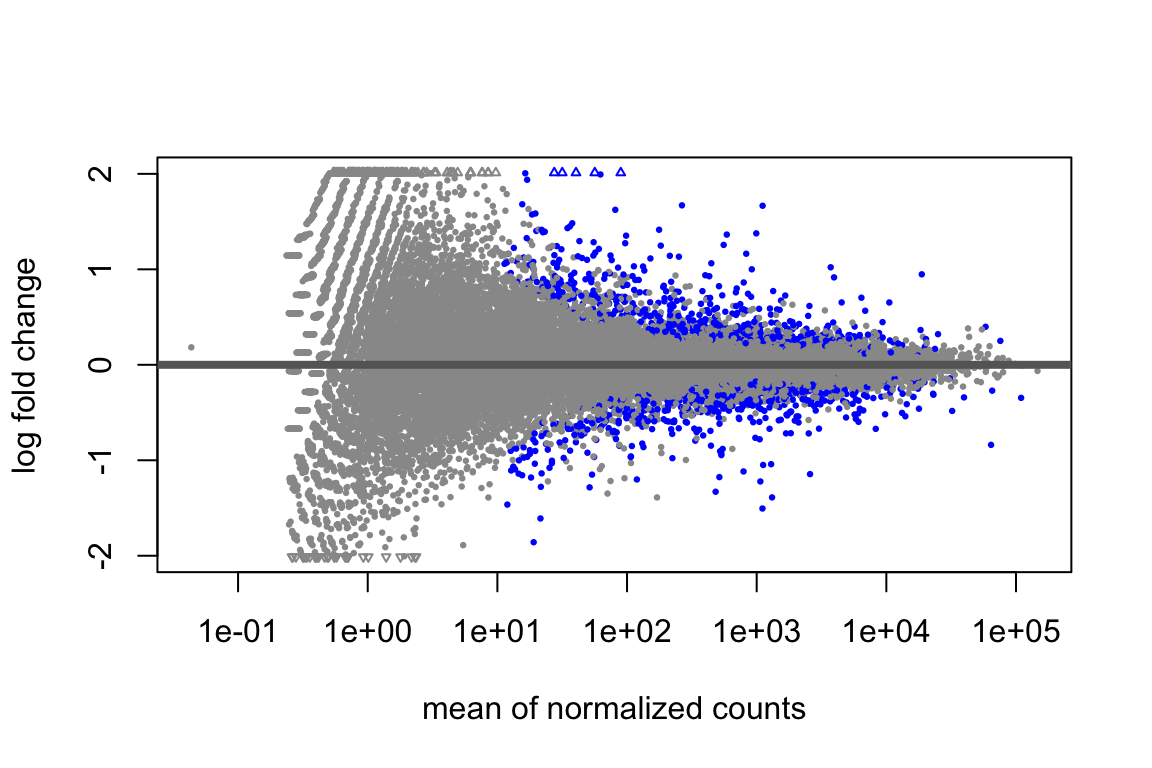
Nell’analisi RNA-seq, lo shrinkage è una tecnica statistica che migliora l’interpretabilità e l’accuratezza dei risultati, soprattutto per i geni con bassa espressione o alta varianza. DESeq2 offre metodi di shrinkage sofisticati per i log2 fold change, che aiutano a ottenere stime più robuste e affidabili.
plotMA()
Prima di applicare lo shrinkage, visualizziamo i risultati grezzi del test di Wald usando la funzione plotMA().
# Crea un MA-plot
plotMA(res)
Questo grafico mostra la relazione tra l’intensità media e il log2 fold change per ogni gene. I geni significativamente differenzialmente espressi sono colorati in blu.
DESeq2 utilizza un metodo di shrinkage bayesiano che “restringe” le stime dei log2 fold change verso valori più probabili, basandosi sulla distribuzione a priori dei log2 fold change e sulla dispersione stimata per ciascun gene. I geni con bassa espressione o alta varianza vengono “shrunk” maggiormente verso zero, mentre i geni con alta espressione e bassa varianza vengono “shrunk” meno.
# Applica lo shrinkage con la funzione lfcShrink()
resultsNames(dds)
#> [1] "Intercept"
#> [2] "infection_NonInfected_vs_InfluenzaA"
resShrink <- lfcShrink(dds, coef = "infection_NonInfected_vs_InfluenzaA", type = "apeglm")
#> using 'apeglm' for LFC shrinkage. If used in published research, please cite:
#> Zhu, A., Ibrahim, J.G., Love, M.I. (2018) Heavy-tailed prior distributions for
#> sequence count data: removing the noise and preserving large differences.
#> Bioinformatics. https://doi.org/10.1093/bioinformatics/bty895In questo esempio, applichiamo lo shrinkage al coefficiente che rappresenta il confronto tra la condizione “trattato” e la condizione “controllo”. Il metodo “apeglm” è un metodo di shrinkage accurato e veloce.
# Crea un MA-plot dopo lo shrinkage
plotMA(resShrink)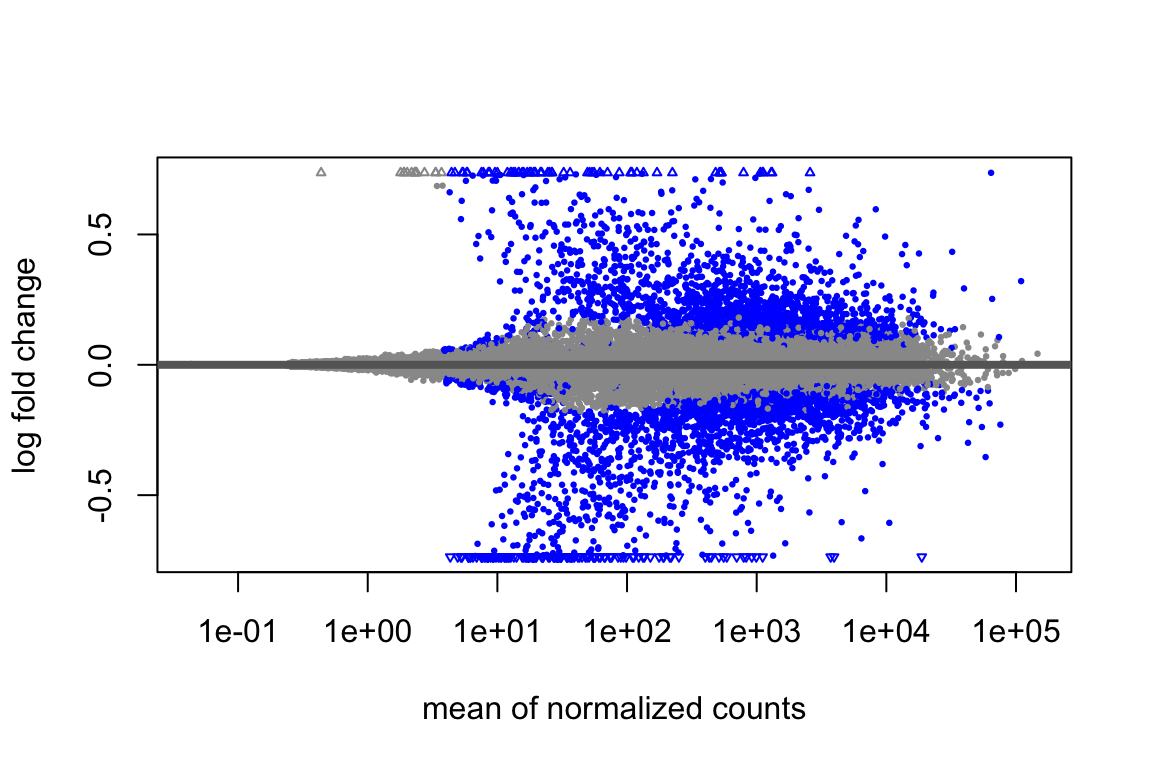
Confrontando questo grafico con il precedente, si può osservare come lo shrinkage abbia moderato i log2 fold change, soprattutto per i geni con bassa espressione.