Il dataset che useremo in questo corso è disponibile presso il Gene Expression Omnibus (GEO), con numero di accesso GSE96870.
1.1 Descrizione del dataset
Stato: Pubblico dal 14 giugno 2017 Titolo: L’effetto dell’infezione delle vie respiratorie superiori sui cambiamenti trascrittomici nel SNC Organismo: Mus musculus (topo domestico) Tipo di esperimento: Profilo di espressione mediante sequenziamento ad alta produttività
Sommario:
Scopo: L’obiettivo di questo studio era determinare l’effetto di un’infezione delle vie respiratorie superiori sui cambiamenti nella trascrizione dell’RNA che si verificano nel cervelletto e nel midollo spinale dopo l’infezione.
Metodi: Topi C57BL/6 di otto settimane di età, appaiati per sesso, sono stati inoculati con soluzione salina o con Influenza A (Puerto Rico/8/34; PR8, 1.0 HAU) per via intranasale e i cambiamenti trascrittomici nei tessuti del cervelletto e del midollo spinale sono stati valutati mediante RNA-seq (letture paired-end da 100 bp) ai giorni 0 (non infetti), 4 e 8.
Risultati: Dopo il trimming e l’esclusione delle letture multi-mapping, una media del 92,07% (cervelletto) e del 91,71% (midollo spinale) dei geni sono stati mappati in modo univoco a un gene. Il numero medio di letture single-end per campione era 36,42 e 37 milioni rispettivamente per il midollo spinale e il cervelletto. L’infezione ha causato cambiamenti significativi al trascrittoma di ciascun tessuto, che sono stati più evidenti al giorno 8 dopo l’infezione.
Conclusione: Questo studio è il primo a utilizzare la tecnologia RNA-seq per valutare l’effetto dell’infezione periferica da influenza A sui cambiamenti nell’espressione genica del cervelletto e del midollo spinale.
Disegno complessivo: Topi C57BL/6 di otto settimane di età, appaiati per sesso, sono stati inoculati con soluzione salina o con Influenza A (Puerto Rico/8/34; PR8, 1.0 HAU) per via intranasale e i cambiamenti trascrittomici nei tessuti del cervelletto e del midollo spinale sono stati valutati mediante RNA-seq (letture paired-end da 100 bp) ai giorni 0 (non infetti), 4 e 8.
1.2 Lista dei campioni
Mostra il codice R
# downloading raw dataif(!fs::dir_exists("data")){fs::dir_create("data")}# The dataset is available at Gene Expression Omnibus (GEO), under the accession number GSE96870. if(!fs::file_exists("data/GSE96870_counts_cerebellum.csv")){download.file( url ="https://github.com/carpentries-incubator/bioc-rnaseq/raw/main/episodes/data/GSE96870_counts_cerebellum.csv", destfile ="data/GSE96870_counts_cerebellum.csv")}if(!fs::file_exists("data/GSE96870_coldata_all.csv")){download.file( url ="https://github.com/carpentries-incubator/bioc-rnaseq/raw/main/episodes/data/GSE96870_coldata_all.csv", destfile ="data/GSE96870_coldata_all.csv")}if(!fs::file_exists("data/GSE96870_rowranges.tsv")){download.file( url ="https://github.com/carpentries-incubator/bioc-rnaseq/raw/main/episodes/data/GSE96870_rowranges.tsv", destfile ="data/GSE96870_rowranges.tsv")}# read all data descriptioncoldata_all<-read_csv("data/GSE96870_coldata_all.csv")datatable(coldata_all|>select(sample, sex, infection, time, tissue), options =list( columnDefs =list(list(className ='dt-center', targets =5)), pageLength =10, lengthMenu =c(10, 20, 30, 40 , 50)))
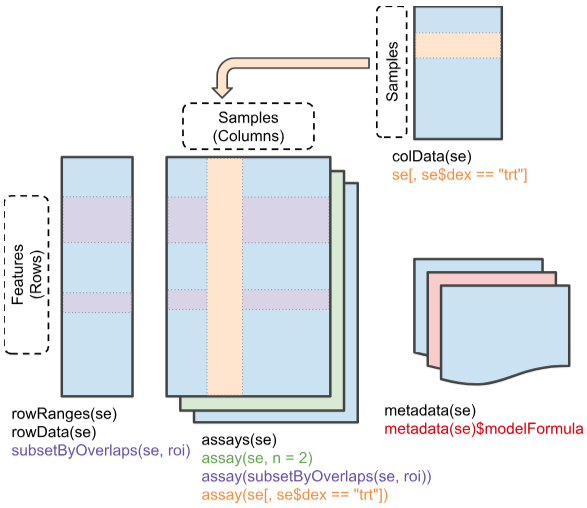
Nel mondo dell’analisi genomica, la gestione e l’organizzazione dei dati sono fondamentali. La classe SummarizedExperiment offre una soluzione robusta ed efficiente per archiviare e manipolare dati provenienti da esperimenti di sequenziamento e microarray, come RNA-Seq e ChIP-Seq.
Immagina un contenitore simile a una matrice in cui:
Le righe rappresentano le caratteristiche di interesse: geni, trascritti, esoni, o qualsiasi altra unità genomica che stai studiando.
Le colonne rappresentano i campioni: individui, tessuti, o condizioni sperimentali diverse.
All’interno di questo contenitore, SummarizedExperiment memorizza non solo i dati grezzi dell’esperimento (ad esempio, il conteggio delle reads per ogni gene in ogni campione), ma anche informazioni cruciali sui campioni e sulle caratteristiche stesse.
Ecco i componenti principali di un SummarizedExperiment:
Saggi (assays): Matrici contenenti i dati quantitativi dell’esperimento. Possono essere presenti più saggi all’interno dello stesso oggetto, ad esempio per memorizzare diverse tipologie di dati omici.
Metadati del campione (colData): Un data.frame che associa a ciascun campione informazioni aggiuntive come età, sesso, condizione di salute, o qualsiasi altra variabile rilevante per l’analisi.
Metadati delle caratteristiche (rowData): Un data.frame che descrive le caratteristiche, fornendo ad esempio ID genetici, annotazioni funzionali, o posizione genomica.
Vantaggi di SummarizedExperiment:
Coordinamento tra dati e metadati:SummarizedExperiment garantisce la sincronizzazione tra i dati sperimentali e i metadati. Se escludi un campione dai dati, le informazioni corrispondenti nei metadati vengono automaticamente rimosse, evitando errori e incongruenze.
Flessibilità:SummarizedExperiment può gestire diverse tipologie di dati e metadati, adattandosi a una vasta gamma di esperimenti genomici.
Efficienza: La struttura organizzata di SummarizedExperiment facilita l’accesso e la manipolazione dei dati, semplificando le analisi a valle.
1.4.1 Esempio:
In un esperimento di RNA-Seq, SummarizedExperiment potrebbe contenere:
Saggio: una matrice con i conteggi di reads per ogni gene (riga) in ciascun campione (colonna).
Metadati del campione: informazioni sul tessuto di origine di ciascun campione (es. cervello, fegato, ecc.).
Metadati delle caratteristiche: annotazioni funzionali dei geni (es. gene codificante per una proteina, gene non codificante, ecc.).
Il pacchetto airway contiene un dataset di esempio proveniente da un esperimento di RNA-Seq che misura il numero di reads per gene in cellule di muscolatura liscia delle vie aeree.
DataFrame with 63677 rows and 10 columns
gene_id gene_name entrezid gene_biotype
<character> <character> <integer> <character>
ENSG00000000003 ENSG00000000003 TSPAN6 NA protein_coding
ENSG00000000005 ENSG00000000005 TNMD NA protein_coding
ENSG00000000419 ENSG00000000419 DPM1 NA protein_coding
ENSG00000000457 ENSG00000000457 SCYL3 NA protein_coding
ENSG00000000460 ENSG00000000460 C1orf112 NA protein_coding
... ... ... ... ...
ENSG00000273489 ENSG00000273489 RP11-180C16.1 NA antisense
ENSG00000273490 ENSG00000273490 TSEN34 NA protein_coding
ENSG00000273491 ENSG00000273491 RP11-138A9.2 NA lincRNA
ENSG00000273492 ENSG00000273492 AP000230.1 NA lincRNA
ENSG00000273493 ENSG00000273493 RP11-80H18.4 NA lincRNA
gene_seq_start gene_seq_end seq_name seq_strand
<integer> <integer> <character> <integer>
ENSG00000000003 99883667 99894988 X -1
ENSG00000000005 99839799 99854882 X 1
ENSG00000000419 49551404 49575092 20 -1
ENSG00000000457 169818772 169863408 1 -1
ENSG00000000460 169631245 169823221 1 1
... ... ... ... ...
ENSG00000273489 131178723 131182453 7 -1
ENSG00000273490 54693789 54697585 HSCHR19LRC_LRC_J_CTG1 1
ENSG00000273491 130600118 130603315 HG1308_PATCH 1
ENSG00000273492 27543189 27589700 21 1
ENSG00000273493 58315692 58315845 3 1
seq_coord_system symbol
<integer> <character>
ENSG00000000003 NA TSPAN6
ENSG00000000005 NA TNMD
ENSG00000000419 NA DPM1
ENSG00000000457 NA SCYL3
ENSG00000000460 NA C1orf112
... ... ...
ENSG00000273489 NA RP11-180C16.1
ENSG00000273490 NA TSEN34
ENSG00000273491 NA RP11-138A9.2
ENSG00000273492 NA AP000230.1
ENSG00000273493 NA RP11-80H18.4
Source Code
---params: mycondition: infection mynum: InfluenzaA mydenom: NonInfected mypval: 0.01 myfc: 0.8 mypadj: fdr---```{r}#| echo: false#| message: false#| warning: falselibrary(tidyverse)library(DESeq2) # BioClibrary(RColorBrewer)library(pheatmap)library(ggrepel)library(cowplot)library(DT)library(scales)library(vsn) # BioClibrary(apeglm) # BioClibrary(rmarkdown)library(gt)```# Importazione dei dati {#sec-preproc-import}::: {.content-hidden unless-meta="features.advanced_analysis"}Il dataset è disponibile presso il Gene Expression Omnibus (GEO): [GSE96870](https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE96870).::::::: {.content-hidden when-meta="features.advanced_analysis"}Il dataset che useremo in questo corso è disponibile presso il Gene Expression Omnibus (GEO), con numero di accesso [GSE96870](https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE96870).## Descrizione del dataset**Stato:** Pubblico dal 14 giugno 2017\**Titolo:** L'effetto dell'infezione delle vie respiratorie superiori sui cambiamenti trascrittomici nel SNC\**Organismo:** Mus musculus (topo domestico)\**Tipo di esperimento:** Profilo di espressione mediante sequenziamento ad alta produttività\**Sommario:**::: {style="margin-left: 20px"}- **Scopo:** L'obiettivo di questo studio era determinare l'effetto di un'infezione delle vie respiratorie superiori sui cambiamenti nella trascrizione dell'RNA che si verificano nel cervelletto e nel midollo spinale dopo l'infezione.\- **Metodi:** Topi C57BL/6 di otto settimane di età, appaiati per sesso, sono stati inoculati con soluzione salina o con Influenza A (Puerto Rico/8/34; PR8, 1.0 HAU) per via intranasale e i cambiamenti trascrittomici nei tessuti del cervelletto e del midollo spinale sono stati valutati mediante RNA-seq (letture paired-end da 100 bp) ai giorni 0 (non infetti), 4 e 8.\- **Risultati:** Dopo il trimming e l'esclusione delle letture multi-mapping, una media del 92,07% (cervelletto) e del 91,71% (midollo spinale) dei geni sono stati mappati in modo univoco a un gene. Il numero medio di letture single-end per campione era 36,42 e 37 milioni rispettivamente per il midollo spinale e il cervelletto. L'infezione ha causato cambiamenti significativi al trascrittoma di ciascun tessuto, che sono stati più evidenti al giorno 8 dopo l'infezione.\- **Conclusione:** Questo studio è il primo a utilizzare la tecnologia RNA-seq per valutare l'effetto dell'infezione periferica da influenza A sui cambiamenti nell'espressione genica del cervelletto e del midollo spinale.\:::**Disegno complessivo:** Topi C57BL/6 di otto settimane di età, appaiati per sesso, sono stati inoculati con soluzione salina o con Influenza A (Puerto Rico/8/34; PR8, 1.0 HAU) per via intranasale e i cambiamenti trascrittomici nei tessuti del cervelletto e del midollo spinale sono stati valutati mediante RNA-seq (letture paired-end da 100 bp) ai giorni 0 (non infetti), 4 e 8.\::::## Lista dei campioni```{r}#| echo: true#| message: false#| warning: false#| tbl-cap: "Tabella con tutti i campioni presenti nell'esperimento"# downloading raw dataif(!fs::dir_exists("data")){ fs::dir_create("data")}# The dataset is available at Gene Expression Omnibus (GEO), under the accession number GSE96870. if(!fs::file_exists("data/GSE96870_counts_cerebellum.csv")){download.file(url ="https://github.com/carpentries-incubator/bioc-rnaseq/raw/main/episodes/data/GSE96870_counts_cerebellum.csv", destfile ="data/GSE96870_counts_cerebellum.csv")}if(!fs::file_exists("data/GSE96870_coldata_all.csv")){download.file(url ="https://github.com/carpentries-incubator/bioc-rnaseq/raw/main/episodes/data/GSE96870_coldata_all.csv", destfile ="data/GSE96870_coldata_all.csv" )}if(!fs::file_exists("data/GSE96870_rowranges.tsv")){download.file(url ="https://github.com/carpentries-incubator/bioc-rnaseq/raw/main/episodes/data/GSE96870_rowranges.tsv", destfile ="data/GSE96870_rowranges.tsv")}# read all data descriptioncoldata_all <-read_csv("data/GSE96870_coldata_all.csv")datatable(coldata_all |>select(sample, sex, infection, time, tissue),options =list(columnDefs =list(list(className ='dt-center', targets =5)),pageLength =10,lengthMenu =c(10, 20, 30, 40 , 50)))```## Summary dei campioni```{r}datatable(coldata_all |> dplyr::count(sex, infection, time, tissue),options =list(columnDefs =list(list(className ='dt-center', targets =5)),pageLength =12))```## Summarized experiment```{r}#| label: Summarized #| echo: false#| out.width: 600knitr::include_graphics("images/summarizedExp.png")```Nel mondo dell'analisi genomica, la gestione e l'organizzazione dei dati sono fondamentali. La classe `SummarizedExperiment` offre una soluzione robusta ed efficiente per archiviare e manipolare dati provenienti da esperimenti di sequenziamento e microarray, come RNA-Seq e ChIP-Seq.Immagina un contenitore simile a una matrice in cui:- **Le righe rappresentano le caratteristiche di interesse:** geni, trascritti, esoni, o qualsiasi altra unità genomica che stai studiando.- **Le colonne rappresentano i campioni:** individui, tessuti, o condizioni sperimentali diverse.All'interno di questo contenitore, `SummarizedExperiment` memorizza non solo i dati grezzi dell'esperimento (ad esempio, il conteggio delle reads per ogni gene in ogni campione), ma anche informazioni cruciali sui campioni e sulle caratteristiche stesse.**Ecco i componenti principali di un `SummarizedExperiment`:**- **Saggi (assays):** Matrici contenenti i dati quantitativi dell'esperimento. Possono essere presenti più saggi all'interno dello stesso oggetto, ad esempio per memorizzare diverse tipologie di dati omici.- **Metadati del campione (colData):** Un `data.frame` che associa a ciascun campione informazioni aggiuntive come età, sesso, condizione di salute, o qualsiasi altra variabile rilevante per l'analisi.- **Metadati delle caratteristiche (rowData):** Un `data.frame` che descrive le caratteristiche, fornendo ad esempio ID genetici, annotazioni funzionali, o posizione genomica.**Vantaggi di `SummarizedExperiment`:**- **Coordinamento tra dati e metadati:** `SummarizedExperiment` garantisce la sincronizzazione tra i dati sperimentali e i metadati. Se escludi un campione dai dati, le informazioni corrispondenti nei metadati vengono automaticamente rimosse, evitando errori e incongruenze.- **Flessibilità:** `SummarizedExperiment` può gestire diverse tipologie di dati e metadati, adattandosi a una vasta gamma di esperimenti genomici.- **Efficienza:** La struttura organizzata di `SummarizedExperiment` facilita l'accesso e la manipolazione dei dati, semplificando le analisi a valle.### Esempio:In un esperimento di RNA-Seq, `SummarizedExperiment` potrebbe contenere:- **Saggio:** una matrice con i conteggi di reads per ogni gene (riga) in ciascun campione (colonna).- **Metadati del campione:** informazioni sul tessuto di origine di ciascun campione (es. cervello, fegato, ecc.).- **Metadati delle caratteristiche:** annotazioni funzionali dei geni (es. gene codificante per una proteina, gene non codificante, ecc.).Il pacchetto `airway` contiene un dataset di esempio proveniente da un esperimento di RNA-Seq che misura il numero di reads per gene in cellule di muscolatura liscia delle vie aeree.```{r}#| echo: true#| message: false#| warning: falselibrary(SummarizedExperiment)data(airway, package="airway")airway```**Saggio:**```{r}assay(airway)[1:20, 1:5]```**Metadati del campione:**```{r}colData(airway)```**Metadati delle caratteristiche:**```{r}rowData(airway)```