DESeq2 utilizza un modello statistico sofisticato per analizzare i dati di conteggio RNA-seq e identificare i geni differenzialmente espressi. Questo modello tiene conto della natura discreta dei dati di conteggio e della variabilità intrinseca degli esperimenti RNA-seq.
Distribuzione Binomiale Negativa
Il modello di DESeq2 si basa sulla distribuzione binomiale negativa, una distribuzione di probabilità discreta che è adatta per modellare dati di conteggio con sovradispersione. La sovradispersione si verifica quando la varianza dei dati è maggiore di quanto ci si aspetterebbe dalla distribuzione di Poisson, che è spesso il caso dei dati RNA-seq.
Formulazione del Modello
Il modello di DESeq2 può essere formulato come segue:
K_ij ~ NB(μ_ij, α_i)
dove:
K_ij è il conteggio del gene i nel campione j
μ_ij è la media del conteggio, che dipende da fattori sperimentali e da altri covariati
α_i è il parametro di dispersione del gene i, che cattura la variabilità tra i replicati
Stima dei Parametri
DESeq2 stima i parametri del modello, inclusi i fattori di dimensione, la dispersione genica e i coefficienti del modello lineare generalizzato (GLM), utilizzando metodi di massima verosimiglianza.
Fasi della Modellazione
Stima dei fattori di dimensione: DESeq2 normalizza i dati di conteggio per tenere conto delle differenze nella dimensione della libreria (visto nel capitolo precedente).
Stima della dispersione: DESeq2 stima la dispersione genica, ovvero la variabilità tra i replicati per ciascun gene.
Adattamento del GLM: DESeq2 adatta un GLM per modellare la relazione tra l’espressione genica e i fattori sperimentali.
Test statistici: DESeq2 applica test statistici (test di Wald) per identificare i geni differenzialmente espressi (prossimo capitolo).
Vantaggi del Modello di DESeq2
Flessibilità: Il modello può essere adattato a diversi disegni sperimentali, inclusi disegni fattoriali, disegni con variabili continue e disegni con interazioni.
Robustezza: Il modello è robusto alla sovradispersione e agli outlier.
Accuratezza: Il modello fornisce stime accurate dell’espressione differenziale.
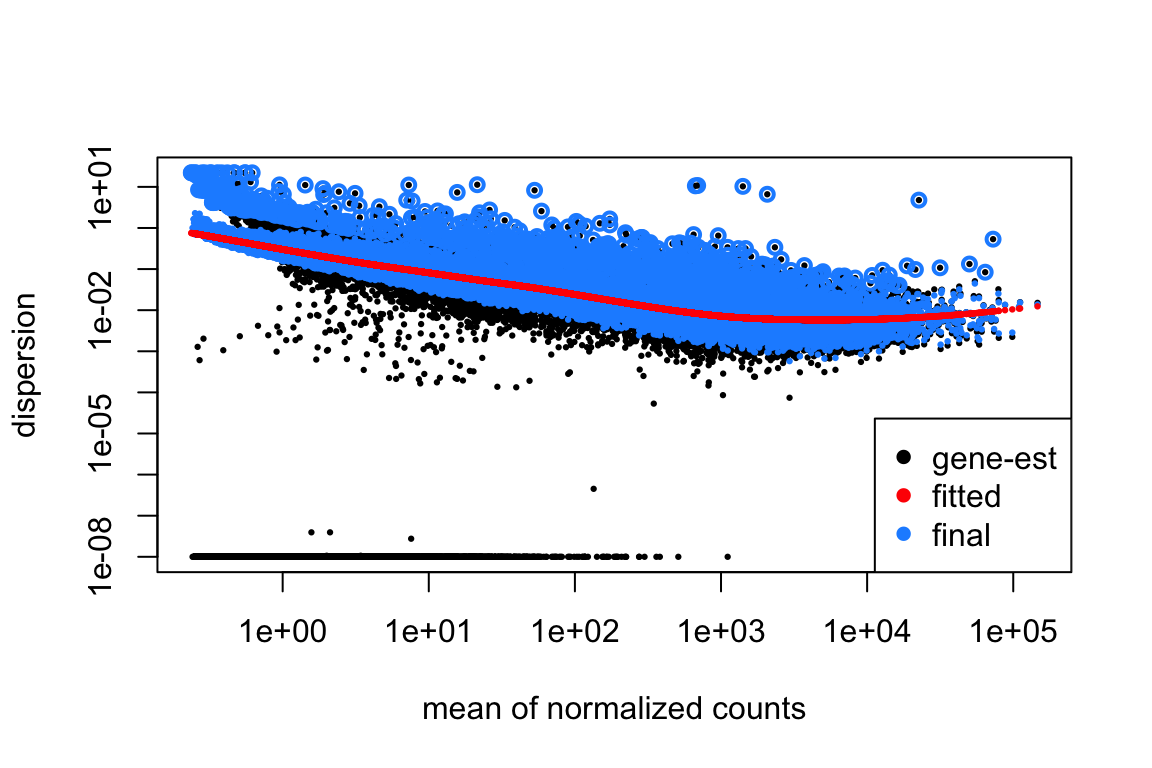
8.1 Dispersion plot
Il grafico a dispersione, visualizzando la relazione tra media normalizzata dei conteggi e dispersione stimata per ciascun gene, consente di valutare la qualità dei dati, l’efficacia del modello di dispersione implementato in DESeq2 e di identificare geni con livelli di variabilità significativamente diversi dalle attese, potenzialmente influenzati da fattori biologici o tecnici di rilievo.
Assi:
Asse x: Rappresenta la media normalizzata dei conteggi per gene. In altre parole, indica quanto un gene è espresso in media, considerando tutti i campioni e dopo aver normalizzato i conteggi per le dimensioni della libreria.
Asse y: Rappresenta la dispersione stimata per gene. La dispersione è una misura della variabilità dell’espressione di un gene tra i replicati.
Punti:
Punti neri: Ogni punto nero rappresenta un gene. La sua posizione sul grafico indica la sua media e la sua dispersione stimata.
Linea rossa: Questa linea rappresenta la curva di tendenza della dispersione in funzione della media. DESeq2 modella la relazione tra media e dispersione per stimare la dispersione dei geni a bassa espressione, che tendono ad avere stime meno affidabili.
Interpretazione:
Geni con alta dispersione: Geni che si trovano al di sopra della curva di tendenza rossa hanno una dispersione maggiore di quanto previsto dal modello. Questo potrebbe indicare che l’espressione di questi geni è influenzata da fattori biologici o tecnici non considerati nel modello.
Geni con bassa dispersione: Geni che si trovano al di sotto della curva di tendenza rossa hanno una dispersione minore del previsto.
Utilità del grafico:
Valutare la qualità dei dati: Il grafico permette di valutare la qualità dei dati e di identificare eventuali problemi di variabilità.
Verificare l’efficacia del modello: Il grafico mostra come il modello di DESeq2 stima la dispersione.
Identificare geni di interesse: Geni con alta dispersione potrebbero essere di particolare interesse per l’analisi, in quanto la loro espressione potrebbe essere influenzata da fattori biologici rilevanti.
---params: mycondition: infection mynum: InfluenzaA mydenom: NonInfected mypval: 0.01 myfc: 0.8 mypadj: fdr---```{r}#| echo: false#| message: false#| warning: falsesource("_common.R")library(tidyverse)library(DESeq2) # BioClibrary(RColorBrewer)library(pheatmap)library(ggrepel)library(cowplot)library(DT)library(scales)library(vsn) # BioClibrary(apeglm) # BioClibrary(rmarkdown)library(gt)readcounts <-readRDS("data/readcounts.rds")coldata <-readRDS("data/coldata.rds")dds <-readRDS("data/dds_fitered.rds")vst <-readRDS("data/vst.rds")dds_esf <-readRDS("data/dss_esf.rds")```# Modellazione statistica {#sec-deg-stat}DESeq2 utilizza un modello statistico sofisticato per analizzare i dati di conteggio RNA-seq e identificare i geni differenzialmente espressi. Questo modello tiene conto della natura discreta dei dati di conteggio e della variabilità intrinseca degli esperimenti RNA-seq.::: {.content-hidden when-meta="features.advanced_analysis"}**Distribuzione Binomiale Negativa**Il modello di DESeq2 si basa sulla **distribuzione binomiale negativa**, una distribuzione di probabilità discreta che è adatta per modellare dati di conteggio con sovradispersione. La sovradispersione si verifica quando la varianza dei dati è maggiore di quanto ci si aspetterebbe dalla distribuzione di Poisson, che è spesso il caso dei dati RNA-seq.**Formulazione del Modello**Il modello di DESeq2 può essere formulato come segue:``` K_ij ~ NB(μ_ij, α_i)```dove:- `K_ij` è il conteggio del gene *i* nel campione *j*- `μ_ij` è la media del conteggio, che dipende da fattori sperimentali e da altri covariati- `α_i` è il parametro di dispersione del gene *i*, che cattura la variabilità tra i replicati**Stima dei Parametri**DESeq2 stima i parametri del modello, inclusi i fattori di dimensione, la dispersione genica e i coefficienti del modello lineare generalizzato (GLM), utilizzando metodi di massima verosimiglianza.**Fasi della Modellazione**1. **Stima dei fattori di dimensione:** DESeq2 normalizza i dati di conteggio per tenere conto delle differenze nella dimensione della libreria (visto nel capitolo precedente).2. **Stima della dispersione:** DESeq2 stima la dispersione genica, ovvero la variabilità tra i replicati per ciascun gene.3. **Adattamento del GLM:** DESeq2 adatta un GLM per modellare la relazione tra l'espressione genica e i fattori sperimentali.4. **Test statistici:** DESeq2 applica test statistici (test di Wald) per identificare i geni differenzialmente espressi (prossimo capitolo).**Vantaggi del Modello di DESeq2**- **Flessibilità:** Il modello può essere adattato a diversi disegni sperimentali, inclusi disegni fattoriali, disegni con variabili continue e disegni con interazioni.- **Robustezza:** Il modello è robusto alla sovradispersione e agli outlier.- **Accuratezza:** Il modello fornisce stime accurate dell'espressione differenziale.:::## Dispersion plotIl grafico a dispersione, visualizzando la relazione tra media normalizzata dei conteggi e dispersione stimata per ciascun gene, consente di valutare la qualità dei dati, l'efficacia del modello di dispersione implementato in DESeq2 e di identificare geni con livelli di variabilità significativamente diversi dalle attese, potenzialmente influenzati da fattori biologici o tecnici di rilievo.::: {.content-hidden when-meta="features.advanced_analysis"}**Assi:**- **Asse x:** Rappresenta la media normalizzata dei conteggi per gene. In altre parole, indica quanto un gene è espresso in media, considerando tutti i campioni e dopo aver normalizzato i conteggi per le dimensioni della libreria.- **Asse y:** Rappresenta la dispersione stimata per gene. La dispersione è una misura della variabilità dell'espressione di un gene tra i replicati.**Punti:**- **Punti neri:** Ogni punto nero rappresenta un gene. La sua posizione sul grafico indica la sua media e la sua dispersione stimata.- **Linea rossa:** Questa linea rappresenta la curva di tendenza della dispersione in funzione della media. DESeq2 modella la relazione tra media e dispersione per stimare la dispersione dei geni a bassa espressione, che tendono ad avere stime meno affidabili.**Interpretazione:**- **Geni con alta dispersione:** Geni che si trovano al di sopra della curva di tendenza rossa hanno una dispersione maggiore di quanto previsto dal modello. Questo potrebbe indicare che l'espressione di questi geni è influenzata da fattori biologici o tecnici non considerati nel modello.- **Geni con bassa dispersione:** Geni che si trovano al di sotto della curva di tendenza rossa hanno una dispersione minore del previsto.**Utilità del grafico:**- **Valutare la qualità dei dati:** Il grafico permette di valutare la qualità dei dati e di identificare eventuali problemi di variabilità.- **Verificare l'efficacia del modello:** Il grafico mostra come il modello di DESeq2 stima la dispersione.- **Identificare geni di interesse:** Geni con alta dispersione potrebbero essere di particolare interesse per l'analisi, in quanto la loro espressione potrebbe essere influenzata da fattori biologici rilevanti.:::```{r}dds_stat <-estimateDispersions(dds_esf, fitType ="local")plotDispEsts(dds_stat)``````{r}#| echo: falsesaveRDS(dds_stat, "data/dds_stat.rds")```